Adaptive
Immunosequencing
Adaptive Immunosequencing
Adaptive Immunosequencing provides a quantitative, end-to-end immunosequencing solution that helps you discover the breadth and depth of the adaptive immune system. From experimental design to publication-ready data, this solution gives you the power to decipher the complexity of the adaptive immune system, providing fundamental insights into the body’s response to disease and therapy at the cellular level.
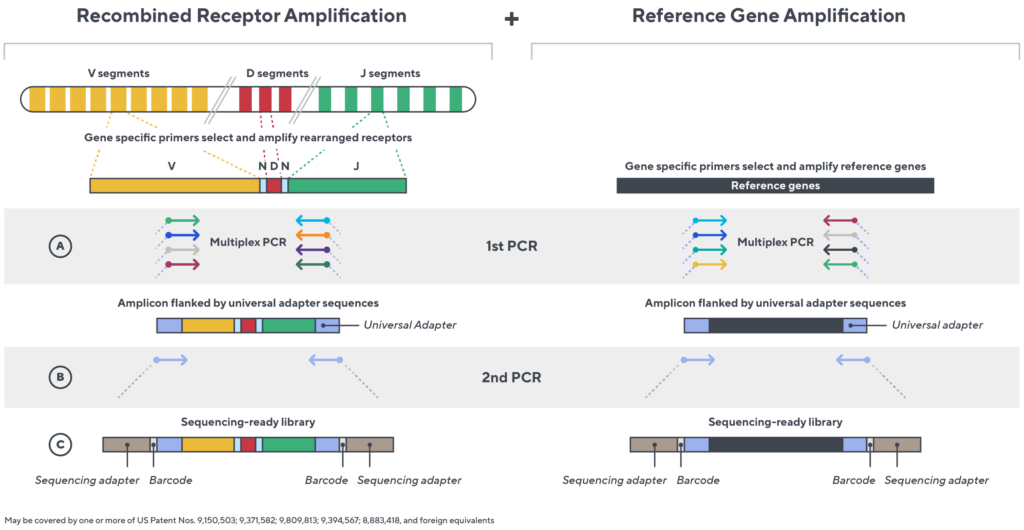
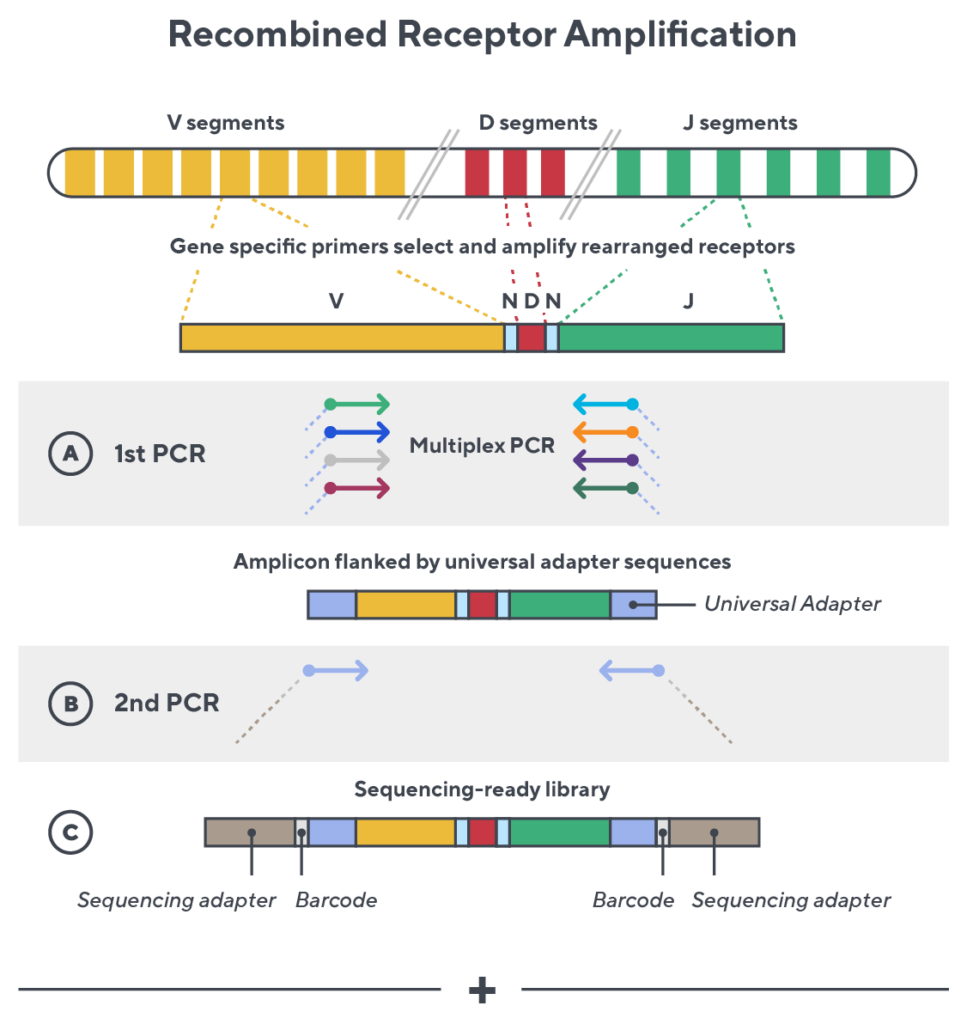
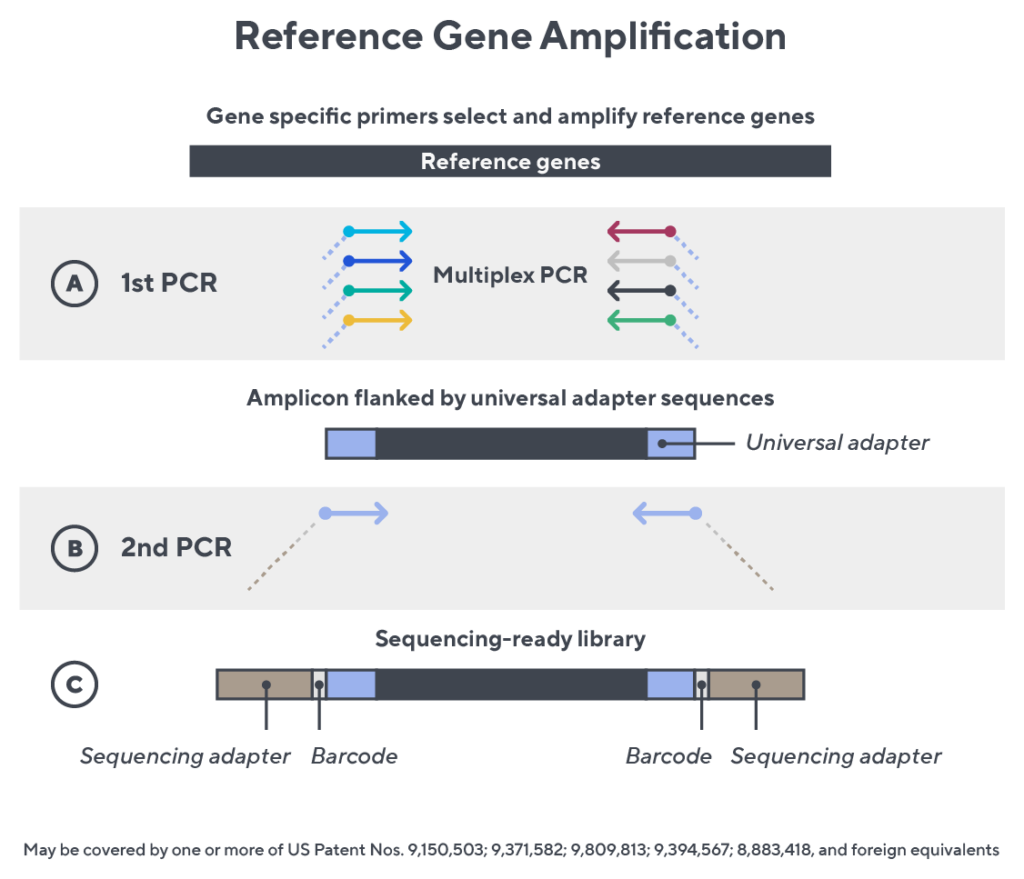
Accuracy and quantification by design
Immunosequencing utilizes a multiplex PCR-based assay enabling sequencing directly from genomic DNA. We provide precise and quantitative abundance data on your cell populations—unlike the relative abundance or variable expression data of RNA-based assays. The assay contains rigorously designed synthetic immune templates as in-line controls plus optimized primers that ensure accurate, quantitative and unbiased results with batch-to-batch consistency.
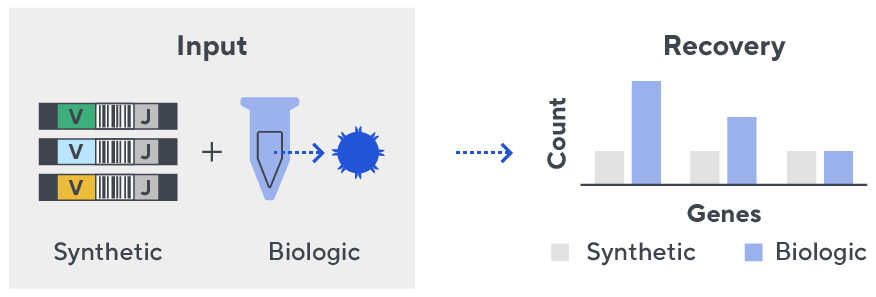
- First, a synthetic immune repertoire was designed to represent all VJ-gene combinations.

- Then, measuring against our synthetic repertoire, we adjusted our primer concentrations to significantly reduce amplification bias.
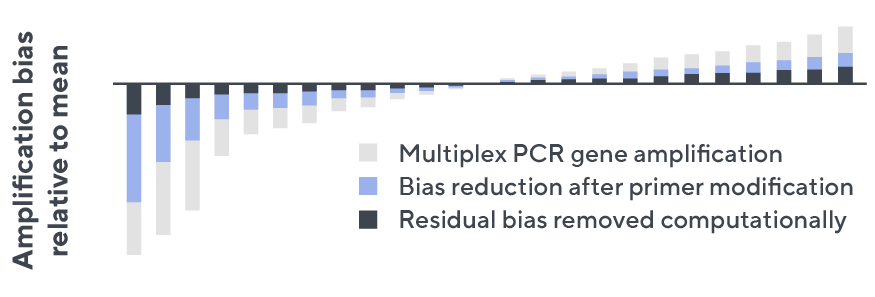
- In-line controls for every sample enable us to measure amplification, correct residual bias, and provide absolute immune cell counts in your sample.
- Our sophisticated bioinformatics platform corrects errors and collapses sequences to deliver highly accurate, high-volume and standardized data sets.

True diversity is about seeing more of the repertoire
Immunosequencing delivers the depth and breadth needed to truly capture the immune repertoire. High-throughput sequencing of the CDR3 variable chain of T- and B-cell receptors provides powerful insights. The CDR3 sequence acts as a unique tag for clonal lineage, enabling tracking of T- and B-cells over time, characterizing the diversity of the immune repertoire and measuring the dynamics of disease and treatment.
Services
Genomic DNA vs. RNA or cDNA
When working with genomic DNA (gDNA) or mRNA, it is important to understand how sample input can impact measurements used to recognize and study the immune repertoire. For archival specimens, gDNA remains more stable compared to mRNA.
With gDNA as starting material, templates can be quantified, providing an absolute cell count. This makes it possible to accurately assess clonal expansion and tissue density of T cells. Complementary DNA (cDNA) or mRNA measurements are obscured by cell expression, because mRNA molecules cannot be directly correlated to cell numbers and may not provide an accurate measure of clonality.
When gDNA is interrogated with an unbiased assay, the output includes:
- Template quantitation
- T-cell fraction
- Absolute cell counts
- Accurate repertoire metrics
HLA classifier
Foreign antigens are presented to T cells by protein complexes called “major histocompatibility complexes” (MHC) that are coded by human leukocyte antigen (HLA) genes. HLA type plays an important role in driving T-cell selection and in shaping T-cell repertoires. The importance of a person’s HLA type in transplant research is well understood, as is the association of specific HLA alleles to common diseases. New research is emerging that explores the link between HLA and disease susceptibility, response to immunotherapy, and more. By adding the HLA classifier to samples processed using Immunosequencing, researchers can unlock new understanding of immune repertoire dynamics in the context of HLA type.
The Immunosequencing assay can infer a sample’s HLA type based on the T-cell receptor (TCR) profile. The HLA classifier uses machine learning to provide a positive or negative call for 145 HLA genes and alleles. This unique tool is based on T-cell receptor sequencing data generated from Adaptive’s partnership with Microsoft and our Antigen Map project from ~4,000 HLA typed samples.
HLA classifier specifications

Adaptive also offers a cytomegalovirus (CMV) classifier and a COVID classifier. Contact us to learn more.
Data output from the HLA classifier
Each of the 145 models are independent and provide a true/false call for that allele based on the number of the relevant TCRs passing that model’s probability cut-off. The classifier model provides two output files. The Summary of Top Calls file lists all the Class I and Class II allele calls for each sample. The Summary of All Results file is a matrix listing all 145 models and the results for each sample indicated by a true/false call.